Exportome-Atlas FAQ
This FAQ explains the main features of Exportome-Atlas and how to interpret the NES-related annotations, scores, plots, and structural views shown on each protein page.
General Questions
Start here for an overview of what the atlas contains and what appears on each protein page.
1. What is Exportome-Atlas?
Exportome-Atlas is a web-based interactive resource for exploring structure-resolved nuclear export signals (NESs) recognized by exportin XPO1 across the human proteome. It is built from AlphaFold 3.0 models of XPO1–RanGTP–RanBP1–cargo complexes for 4,208 human proteins, identifying 2,961 high-confidence NESs with scores ≥12.
2. What kind of data does Exportome-Atlas provide?
The atlas provides structure-based NES predictions, including:
- composite NES Scores (5–21)
- pocket occupancy patterns (P0–P4)
- NES Classes (including canonical, reverse-orientation, and non-canonical classes)
- sequence context, including domains (SMART/TED) and disorder (MetaPredict)
- juxtaposed NLS annotations
- cofactor-dependent models for proteins where cofactor binding may affect NES geometry
3. Which organisms are currently included?
The current release focuses on the human proteome. It includes 4,208 proteins curated from BioGRID XPO1 interactors, HPA/UniProt nucleo-cytoplasmic shuttling proteins, and disease- or pathway-relevant cargos such as kinases and E3 ligases.
4. How should I cite Exportome-Atlas?
Please cite the associated paper for the current release. If the manuscript is not yet published, this section can be updated with the final citation once available.
5. How do I search for a protein?
You can search by gene symbol (for example, TP53 or PDPK1), protein name (for example, p53), or UniProt ID (for example, P04637) using the landing-page search bar. Search results open protein-specific pages summarizing the predicted NESs for that protein.
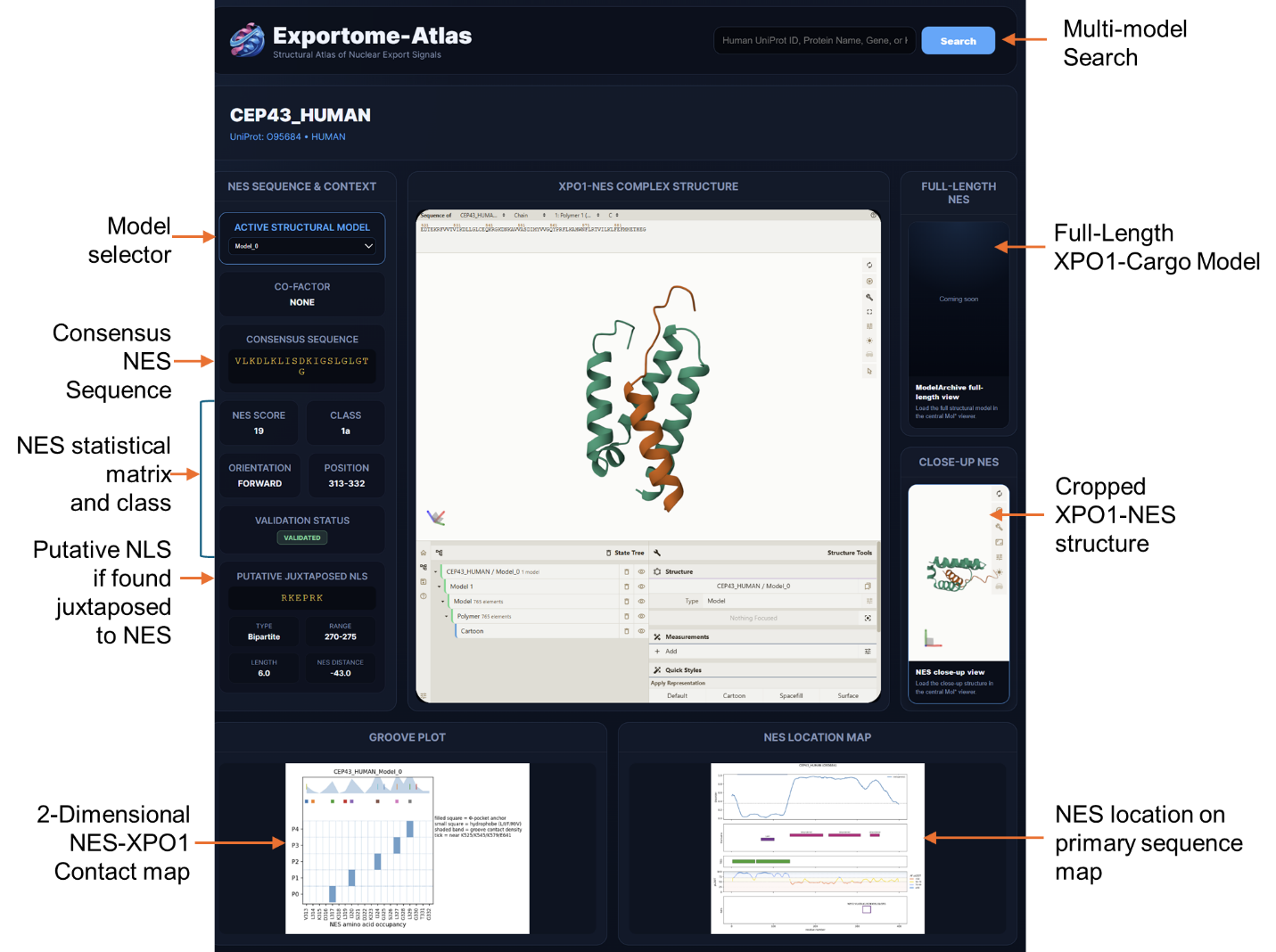
6. What information is shown on a protein page?
Protein pages display:
- identifiers and summary information
- an NES summary table with score, class, position, orientation, cofactor, and validation status
- a Groove Plot
- a 3D molecular viewer of the export complex
- an NES Plot
- a Putative Juxtaposed NLS section, where applicable
7. Can I download data or figures?
Exportome-Atlas is designed to support both exploration and downstream analysis.
Full-data download capability is coming soon and will include:
- NES tables in CSV format
- Groove Plots and NES Plots in publication-ready image formats
- protein summary data
- 3D structures of XPO1–RanGTP–RanBP1–cargo complexes through the molecular viewer
Exact download options may vary by page or release version.
8. Can I report bugs or request new features?
Yes. The website should provide a contact link or support email for reporting issues, suggesting improvements, or requesting features. User feedback is an important part of improving future releases.
9. Are all predicted NESs experimentally validated?
No. Exportome-Atlas includes both experimentally supported NESs and a much larger set of high-confidence structural predictions. Many known cargos are used as benchmarks, but most newly identified NESs have not yet been tested directly in cells. The atlas should therefore be used as both a reference resource and a hypothesis-generation platform.
About the Dataset
These entries explain how to interpret the fields and figures shown for each predicted NES.
1. What does the Consensus Sequence mean?
The Consensus Sequence is the primary NES chosen as the reference for a given protein. It is derived from multiple structural models and represents the region most consistently predicted to occupy the XPO1 groove. Although individual models may vary slightly in their start or end positions, the consensus captures the stable core of the motif. When interpreting NES scores, classes, Groove Plots, or NES Plots, this is the main sequence to focus on.
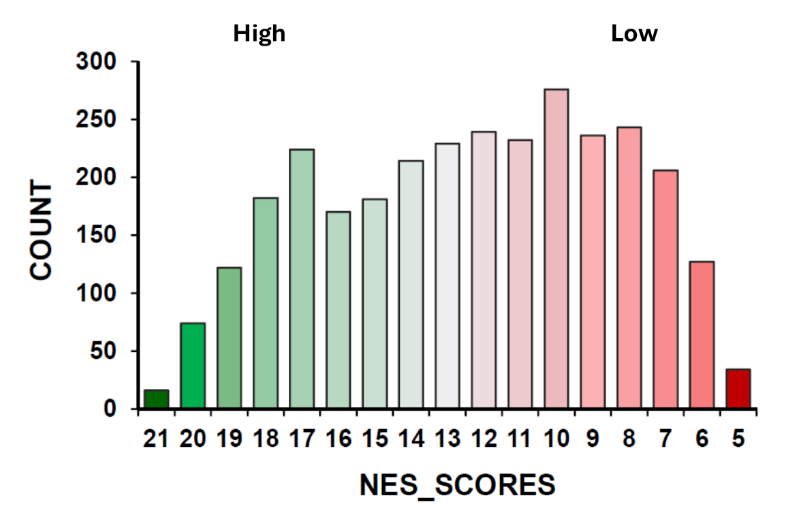
2. What is the NES Score?
The NES Score is a composite measure of the structural support for a predicted NES in a protein. In the current implementation, scores range from 5 (very low confidence) to 21 (very high confidence).
The score integrates several model-derived features, including:
- Model-to-model reproducibility (0–5 points): how consistently the same NES window is recovered across up to five AF3.0 models
- Pocket engagement in the H11–H12 groove (0–5 points + 1 bonus if all 5 anchors land): how many XPO1 pockets (P0–P4) are occupied, and how consistently
- Gatekeeper lysine and groove-rim contacts (0–4 points): hydrogen bonds and polar interactions with key groove residues such as K579 (human K568)
- NES geometry and structural organization (0–6 points): whether the motif adopts a stable, groove-seated amphipathic helix or another compatible conformation
In general, scores from 10–21 indicate progressively stronger support, and scores ≥12 reliably identify export-competent motifs that are reproducible across models. NES Score should always be interpreted alongside the structural model, NES Class, pocket usage, and Validation Status.
3. What does the NES Class mean?
The NES Class describes how a motif engages the XPO1 groove in three dimensions, rather than only by its linear hydrophobic pattern. Classes capture:
- which hydrophobic pockets (P0–P4) are occupied
- the number of residues between anchor positions
- whether the motif binds in the forward (plus) or reverse (minus) orientation
This structure-aware classification refines earlier sequence-based schemes and makes it possible to compare canonical and non-canonical NESs consistently across proteins.
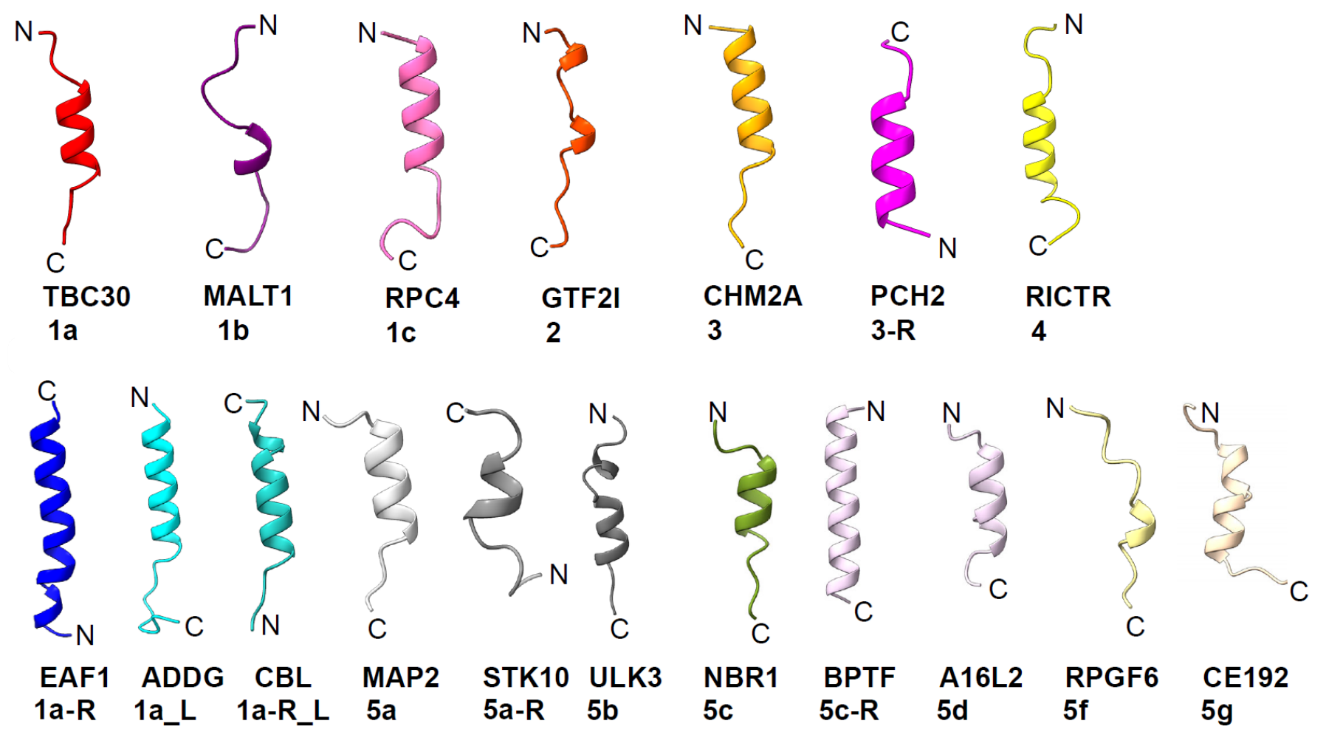
4. What do the NES classes 1a, 1b, 1c, 1d, 2, 3, 4, 5, and undefined mean?
NES classes differ in how many hydrophobic anchors they use and how those anchors are arranged across pockets P0–P4.
- Classes 1a, 1b, 1c, 1d, 2, and 4 are five-anchor NESs spanning P0→P4, each with a distinct inter-anchor spacing pattern
- Class 3 is typically a four-anchor NES engaging P0→P3
- Class 5 includes non-canonical four-anchor NESs with an internal weak or unused pocket and downstream re-engagement, producing subclasses such as 5a–5g
- Undefined is when NES uses anchor residue type or spacing that is not consistent with Class 1-5
Reverse-orientation counterparts (for example, 1a-R, 2-R, 5a-R) follow the same pocketing logic in the opposite direction. Exportome-Atlas assigns these classes using a structure-based classifier derived from Groove Plots and pocket maps.
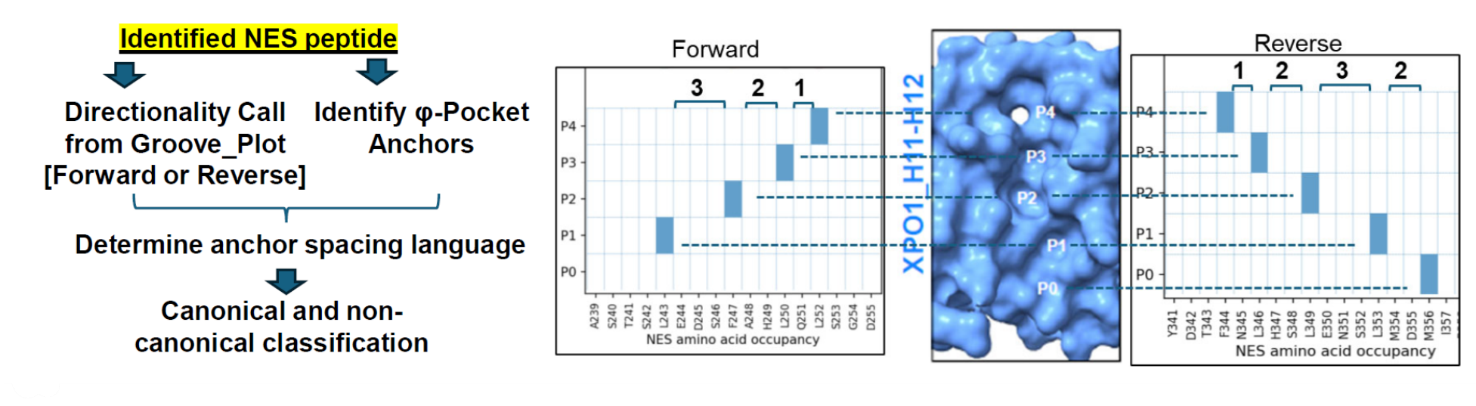
5. What does Orientation mean?
Orientation indicates the direction in which the NES is bound in the XPO1 groove. In plus orientation, the motif follows the canonical pocket order from P0 to P4. In minus orientation, it occupies the same groove in the reverse direction. Because XPO1 can accommodate both modes, orientation is an important part of structural interpretation.
6. What does Position mean?
Position gives the residue range of the NES within the full-length protein. For example, a position of 210–228 means the motif spans residues 210 through 228.
This range is used in NES Plot and domain maps to place the NES relative to:
- annotated domains (SMART/TED)
- intrinsically disordered regions (MetaPredict)
- structural confidence scores (pLDDT) from AlphaFold models generated without XPO1
Position is especially useful for designing mutants, truncations, and export-reporter constructs.
7. What does Cofactor mean?
Cofactor indicates whether a structural model was generated in the presence or absence of a relevant cofactor, such as Zn for zinc-binding proteins. In some cases, the presence or absence of a cofactor can influence local structure and affect how the NES is presented to the XPO1 groove.
8. What does Validation Status mean?
Validation Status shows whether the NES has supporting experimental or curated evidence in the current release.
- Validated entries are supported by experimental data, such as reporter assays, mutational analysis, or literature-curated evidence
- Pending or unvalidated entries are high-confidence structural predictions that have not yet been linked to release-ready validation data
This information can help users prioritize candidates for follow-up experiments.
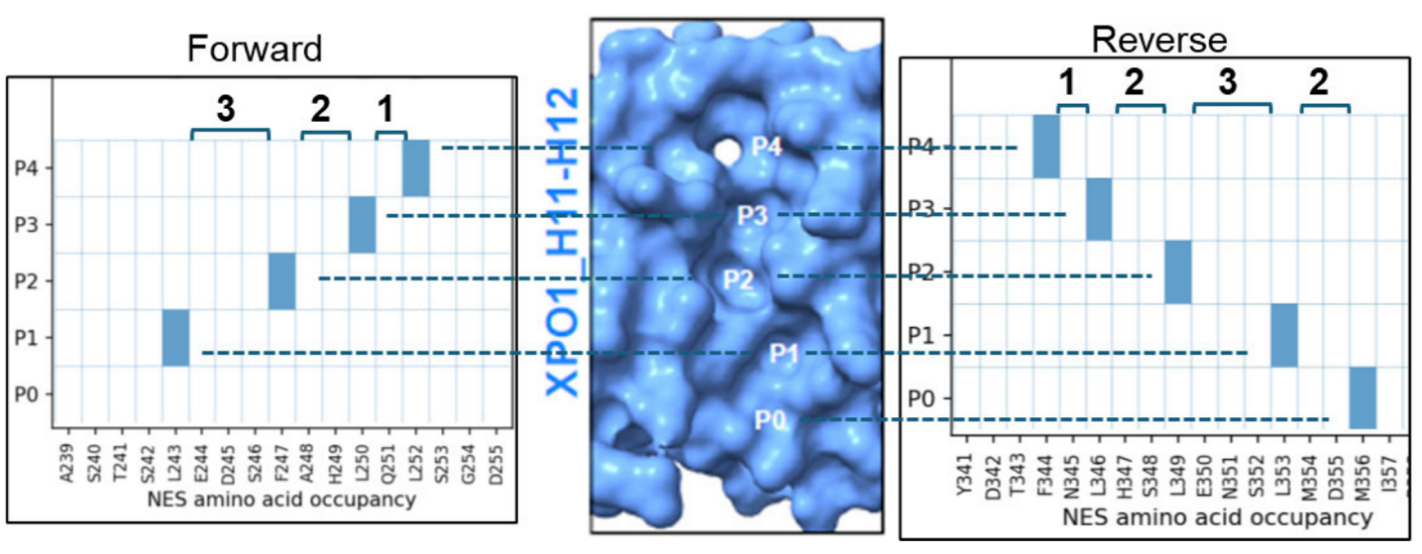
9. What is the Groove Plot?
The Groove Plot is a 2D contact map showing how a given NES engages the XPO1 H11–H12 groove in a specific AF3.0 model. It summarizes which NES residues occupy pockets P0–P4 and makes the anchor pattern easy to read at a glance.
Groove Plots help users:
- identify pocket anchors
- read inter-anchor spacing directly from structural models
- distinguish forward and reverse binding orientations
- compare canonical and non-canonical binding patterns across proteins
If no Groove Plot is shown, the page typically displays a message indicating that the NES is low-scoring or not reliably resolved.
10. What is the NES Plot?
The NES Plot is a sequence-level visualization showing where the NES lies within the full protein and how it relates to surrounding sequence features. It integrates:
- NES position and consensus window
- model-derived NES signal along the sequence
- additional tracks such as intrinsic disorder (MetaPredict), domain annotations (SMART/TED), and pLDDT scores from AlphaFold models without XPO1
This plot helps users assess sequence context, local structural environment, and proximity to domains or regulatory regions. NES Plot should be interpreted together with NES Score, NES Class, and structural models rather than on its own.
11. Why do some entries show “Low-scoring NES or no reliable NES found”?
This message appears when Exportome-Atlas cannot generate a robust structural visualization for the NES in a given entry. Common reasons include:
- a composite NES Score below visualization thresholds
- weak or inconsistent pocket occupancy across models
- failure to form a stable groove-seated helix suitable for Groove Plot extraction
These proteins are still shown for completeness, but the NES should be treated as low confidence and interpreted cautiously.
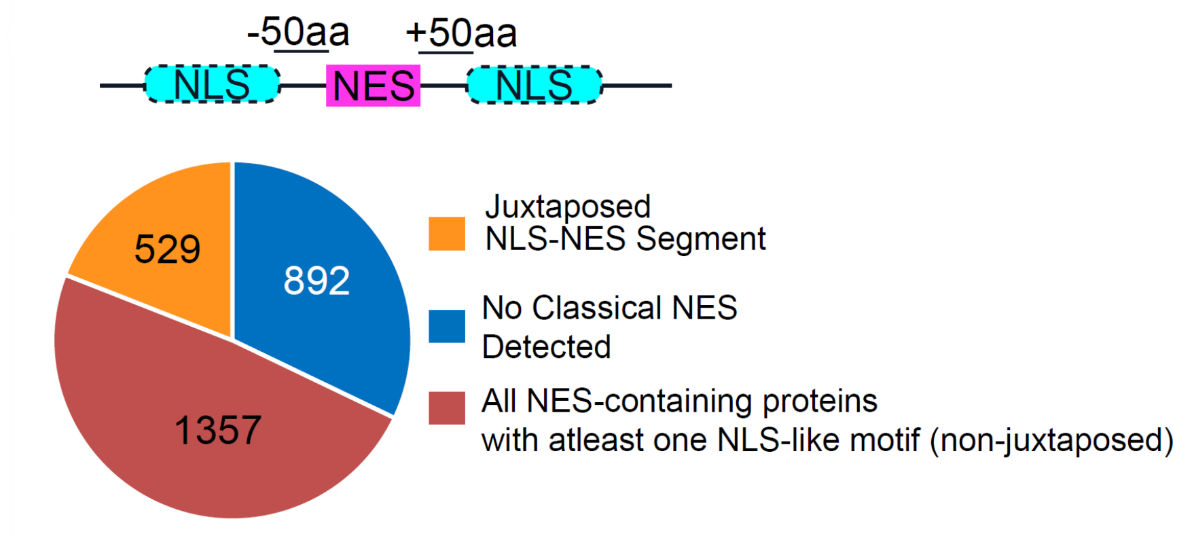
12. What is the Putative Juxtaposed NLS section?
The Putative Juxtaposed NLS section lists nuclear localization signals (NLSs) that occur near the NES in the primary sequence. For each NLS, the atlas may report:
- motif type (for example, monopartite or bipartite)
- sequence and length
- residue range
- distance from the NES
Proteome-wide analysis shows that among 2,778 NES-containing proteins, 529 (about 19%) have at least one classical NLS within ±50 residues of the AF3.0-predicted NES, while 1,357 additional cargos carry an NLS elsewhere in the protein. These arrangements may mark proteins with coordinated import/export control and highlight candidates likely to function as regulated nucleo-cytoplasmic shuttles.
13. How should I interpret model switching?
Each protein entry may include multiple AF3.0 models per condition, with up to five models per cofactor state. Model switching allows you to compare these independent predictions.
- the consensus NES sequence and top-level NES Score usually remain the same, because they are derived from the full ensemble
- model-level features such as Groove Plot, 3D conformation, cofactor state, and fine details of pocket engagement may change
Consistency across models supports a robust prediction, while variability may indicate conformational flexibility or context-dependent NES engagement.
Contact
Exportome Atlas is a companion web resource for exploring structure-resolved nuclear export signals (NESs) and juxtaposed nuclear localization signals (NLSs) across the human exportome.
For questions, bug reports, feature requests, or data inquiries, please contact Chintan Kikani.
Citation
If you use Exportome Atlas in your work, please cite the accompanying manuscript:
AUTHOR LIST. TITLE. bioRxiv (YEAR).
Portal: nuclear.exportome.org
Resource Availability
Exportome Atlas is available at https://www.nuclear.exportome.org. The portal accompanies the current preprint release and may be updated in future versions.